概要
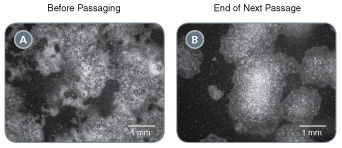
ReLeSR™ is an enzyme-free reagent for dissociation and passaging of human embryonic stem (ES) or induced pluripotent stem (iPS) cells as aggregates without manual selection or scraping. Passaging human ES/iPS cells with ReLeSR™ easily generates optimally-sized aggregates, while eliminating the hassle and variability associated with manual manipulation. By eliminating the need for scraping, ReLeSR™ enables the use of culture flasks and other closed vessels, thus facilitating culture scale-up and automation. ReLeSR™, 500 mL, is now also manufactured following relevant cGMPs under a certified quality management system to ensure the highest quality and consistency for reproducible results.
数据及文献
Publications (12)
Stem cell reports 2019
Pluripotent Stem Cell-Derived Cerebral Organoids Reveal Human Oligodendrogenesis with Dorsal and Ventral Origins.
H. Kim et al.
Abstract
The process of oligodendrogenesis has been relatively well delineated in the rodent brain. However, it remains unknown whether analogous developmental processes are manifested in the human brain. Here we report oligodendrogenesis in forebrain organoids, generated by using OLIG2-GFP knockin human pluripotent stem cell (hPSC) reporter lines. OLIG2/GFP exhibits distinct temporal expression patterns in ventral forebrain organoids (VFOs) versus dorsal forebrain organoids (DFOs). Interestingly, oligodendrogenesis can be induced in both VFOs and DFOs after neuronal maturation. Assembling VFOs and DFOs to generate fused forebrain organoids (FFOs) promotes oligodendroglia maturation. Furthermore, dorsally derived oligodendroglial cells outcompete ventrally derived oligodendroglia and become dominant in FFOs after long-term culture. Thus, our organoid models reveal human oligodendrogenesis with ventral and dorsal origins. These models will serve to study the phenotypic and functional differences between human ventrally and dorsally derived oligodendroglia and to reveal mechanisms of diseases associated with cortical myelin defects.
Stem cell reports 2018 JUL
Disruption of GRIN2B Impairs Differentiation in Human Neurons.
S. Bell et al.
Abstract
Heterozygous loss-of-function mutations in GRIN2B, a subunit of the NMDA receptor, cause intellectual disability and language impairment. We developed clonal models of GRIN2B deletion and loss-of-function mutations in a region coding for the glutamate binding domain in human cells and generated neurons from a patient harboring a missense mutation in the same domain. Transcriptome analysis revealed extensive increases in genes associated with cell proliferation and decreases in genes associated with neuron differentiation, a result supported by extensive protein analyses. Using electrophysiology and calcium imaging, we demonstrate that NMDA receptors are present on neural progenitor cells and that human mutations in GRIN2B can impair calcium influx and membrane depolarization even in a presumed undifferentiated cell state, highlighting an important role for non-synaptic NMDA receptors. It may be this function, in part, which underlies the neurological disease observed in patients with GRIN2B mutations.
Cell 2018 JAN
Intrinsic Immunity Shapes Viral Resistance of Stem Cells.
Wu X et al.
Abstract
Stem cells are highly resistant to viral infection compared to their differentiated progeny; however, the mechanism is mysterious. Here, we analyzed gene expression in mammalian stem cells and cells at various stages of differentiation. We find that, conserved across species, stem cells express a subset of genes previously classified as interferon (IFN) stimulated genes (ISGs) but that expression is intrinsic, as stem cells are refractory to interferon. This intrinsic ISG expression varies in a cell-type-specific manner, and many ISGs decrease upon differentiation, at which time cells become IFN responsive, allowing induction of a broad spectrum of ISGs by IFN signaling. Importantly, we show that intrinsically expressed ISGs protect stem cells against viral infection. We demonstrate the in vivo importance of intrinsic ISG expression for protecting stem cells and their differentiation potential during viral infection. These findings have intriguing implications for understanding stem cell biology and the evolution of pathogen resistance.
Cell stem cell 2017 APR
Human iPSC-Derived Cerebral Organoids Model Cellular Features of Lissencephaly and Reveal Prolonged Mitosis of Outer Radial Glia.
Bershteyn M et al.
Abstract
Classical lissencephaly is a genetic neurological disorder associated with mental retardation and intractable epilepsy, and Miller-Dieker syndrome (MDS) is the most severe form of the disease. In this study, to investigate the effects of MDS on human progenitor subtypes that control neuronal output and influence brain topology, we analyzed cerebral organoids derived from control and MDS-induced pluripotent stem cells (iPSCs) using time-lapse imaging, immunostaining, and single-cell RNA sequencing. We saw a cell migration defect that was rescued when we corrected the MDS causative chromosomal deletion and severe apoptosis of the founder neuroepithelial stem cells, accompanied by increased horizontal cell divisions. We also identified a mitotic defect in outer radial glia, a progenitor subtype that is largely absent from lissencephalic rodents but critical for human neocortical expansion. Our study, therefore, deepens our understanding of MDS cellular pathogenesis and highlights the broad utility of cerebral organoids for modeling human neurodevelopmental disorders.
EMBO molecular medicine 2016 OCT
Coenzyme A corrects pathological defects in human neurons of PANK2-associated neurodegeneration.
Orellana DI et al.
Abstract
Pantothenate kinase-associated neurodegeneration (PKAN) is an early onset and severely disabling neurodegenerative disease for which no therapy is available. PKAN is caused by mutations in PANK2, which encodes for the mitochondrial enzyme pantothenate kinase 2. Its function is to catalyze the first limiting step of Coenzyme A (CoA) biosynthesis. We generated induced pluripotent stem cells from PKAN patients and showed that their derived neurons exhibited premature death, increased ROS production, mitochondrial dysfunctions-including impairment of mitochondrial iron-dependent biosynthesis-and major membrane excitability defects. CoA supplementation prevented neuronal death and ROS formation by restoring mitochondrial and neuronal functionality. Our findings provide direct evidence that PANK2 malfunctioning is responsible for abnormal phenotypes in human neuronal cells and indicate CoA treatment as a possible therapeutic intervention.
Cell reports 2016 NOV
Nucleosome Density ChIP-Seq Identifies Distinct Chromatin Modification Signatures Associated with MNase Accessibility.
Lorzadeh A et al.
Abstract
Nucleosome position, density, and post-translational modification are widely accepted components of mechanisms regulating DNA transcription but still incompletely understood. We present a modified native ChIP-seq method combined with an analytical framework that allows MNase accessibility to be integrated with histone modification profiles. Application of this methodology to the primitive (CD34+) subset of normal human cord blood cells enabled genomic regions enriched in one versus two nucleosomes marked by histone 3 lysine 4 trimethylation (H3K4me3) and/or histone 3 lysine 27 trimethylation (H3K27me3) to be associated with their transcriptional and DNA methylation states. From this analysis, we defined four classes of promoter-specific profiles and demonstrated that a majority of bivalent marked promoters are heterogeneously marked at a single-cell level in this primitive cell type. Interestingly, extension of this approach to human embryonic stem cells revealed an altered relationship between chromatin modification state and nucleosome content at promoters, suggesting developmental stage-specific organization of histone methylation states.
View All Publications
 网站首页
网站首页