 网站首页
网站首页


概要
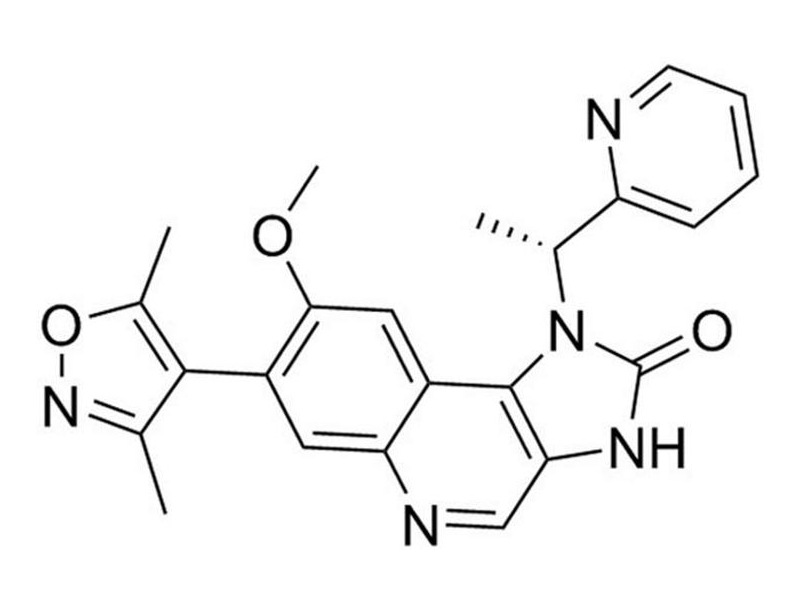
I-BET151 is an inhibitor of bromodomain and extra terminal (BET) family proteins. BET proteins recognize acetylated lysine residues via their 2 bromodomains (Gallenkamp et al.). I-BET151 inhibits BRD2, BRD3, and BRD4 with IC₅₀ values of 0.5, 0.25, and 0.79 µM, respectively (Kline et al.; Vidler et al.; Hewings et al.; Dawson et al. 2012).
REPROGRAMMING
· Enhances reprogramming of mouse fibroblasts to neurons, in combination with ISX-9, Forskolin, and CHIR99021 (Li et al.)
CANCER RESEARCH
· Induces early cell cycle arrest and apoptosis in human and mouse MLL-fusion leukemia cell lines by blocking transcription of key genes including BCL2, C-MYC, and CDK6 (Dawson et al. 2011).
REPROGRAMMING
· Enhances reprogramming of mouse fibroblasts to neurons, in combination with ISX-9, Forskolin, and CHIR99021 (Li et al.)
CANCER RESEARCH
· Induces early cell cycle arrest and apoptosis in human and mouse MLL-fusion leukemia cell lines by blocking transcription of key genes including BCL2, C-MYC, and CDK6 (Dawson et al. 2011).
技术资料
| Document Type | 产品名称 | Catalog # | Lot # | 语言 |
|---|---|---|---|---|
| Product Information Sheet | I-BET151 | 73712, 73714 | All | English |
| Safety Data Sheet | I-BET151 | 73712, 73714 | All | English |
数据及文献
Publications (7)
Cell stem cell 2015 AUG
Small-Molecule-Driven Direct Reprogramming of Mouse Fibroblasts into Functional Neurons.
Abstract
Abstract
Recently, direct reprogramming between divergent lineages has been achieved by the introduction of regulatory transcription factors. This approach may provide alternative cell resources for drug discovery and regenerative medicine, but applications could be limited by the genetic manipulation involved. Here, we show that mouse fibroblasts can be directly converted into neuronal cells using only a cocktail of small molecules, with a yield of up to textgreater90% being TUJ1-positive after 16 days of induction. After a further maturation stage, these chemically induced neurons (CiNs) possessed neuron-specific expression patterns, generated action potentials, and formed functional synapses. Mechanistically, we found that a BET family bromodomain inhibitor, I-BET151, disrupted the fibroblast-specific program, while the neurogenesis inducer ISX9 was necessary to activate neuron-specific genes. Overall, our findings provide a proof of principle" for chemically induced direct reprogramming of somatic cell fates across germ layers without genetic manipulation�
ChemMedChem 2014 MAR
Bromodomains and their pharmacological inhibitors.
Abstract
Abstract
Over 60 bromodomains belonging to proteins with very different functions have been identified in humans. Several of them interact with acetylated lysine residues, leading to the recruitment and stabilization of protein complexes. The bromodomain and extra-terminal domain (BET) proteins contain tandem bromodomains which bind to acetylated histones and are thereby implicated in a number of DNA-centered processes, including the regulation of gene expression. The recent identification of inhibitors of BET and non-BET bromodomains is one of the few examples in which effective blockade of a protein-protein interaction can be achieved with a small molecule. This has led to major strides in the understanding of the function of bromodomain-containing proteins and their involvement in diseases such as cancer and inflammation. Indeed, BET bromodomain inhibitors are now being clinically evaluated for the treatment of hematological tumors and have also been tested in clinical trials for the relatively rare BRD-NUT midline carcinoma. This review gives an overview of the newest developments in the field, with a focus on the biology of selected bromodomain proteins on the one hand, and on reported pharmacological inhibitors on the other, including recent examples from the patent literature.
Journal of medicinal chemistry 2013 APR
Optimization of 3,5-dimethylisoxazole derivatives as potent bromodomain ligands.
Abstract
Abstract
The bromodomain protein module, which binds to acetylated lysine, is emerging as an important epigenetic therapeutic target. We report the structure-guided optimization of 3,5-dimethylisoxazole derivatives to develop potent inhibitors of the BET (bromodomain and extra terminal domain) bromodomain family with good ligand efficiency. X-ray crystal structures of the most potent compounds reveal key interactions required for high affinity at BRD4(1). Cellular studies demonstrate that the phenol and acetate derivatives of the lead compounds showed strong antiproliferative effects on MV4;11 acute myeloid leukemia cells, as shown for other BET bromodomain inhibitors and genetic BRD4 knockdown, whereas the reported compounds showed no general cytotoxicity in other cancer cell lines tested.
Journal of medicinal chemistry 2012 SEP
Druggability analysis and structural classification of bromodomain acetyl-lysine binding sites.
Abstract
Abstract
Bromodomains are readers of the epigenetic code that specifically bind acetyl-lysine containing recognition sites on proteins. Recently the BET family of bromodomains has been demonstrated to be druggable through the discovery of potent inhibitors, sparking an interest in protein-protein interaction inhibitors that directly target gene transcription. Here, we assess the druggability of diverse members of the bromodomain family using SiteMap and show that there are significant differences in predicted druggability. Furthermore, we trace these differences in druggability back to unique amino acid signatures in the bromodomain acetyl-lysine binding sites. These signatures were then used to generate a new classification of the bromodomain family, visualized as a classification tree. This represents the first analysis of this type for the bromodomain family and can prove useful in the discovery of inhibitors, particularly for anticipating screening hit rates, identifying inhibitors that can be explored for lead hopping approaches, and selecting proteins for selectivity screening.
The New England journal of medicine 2012 AUG
Targeting epigenetic readers in cancer.
Abstract
Abstract
Nature 2011 OCT
Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia.
Abstract
Abstract
Recurrent chromosomal translocations involving the mixed lineage leukaemia (MLL) gene initiate aggressive forms of leukaemia, which are often refractory to conventional therapies. Many MLL-fusion partners are members of the super elongation complex (SEC), a critical regulator of transcriptional elongation, suggesting that aberrant control of this process has an important role in leukaemia induction. Here we use a global proteomic strategy to demonstrate that MLL fusions, as part of SEC and the polymerase-associated factor complex (PAFc), are associated with the BET family of acetyl-lysine recognizing, chromatin 'adaptor' proteins. These data provided the basis for therapeutic intervention in MLL-fusion leukaemia, via the displacement of the BET family of proteins from chromatin. We show that a novel small molecule inhibitor of the BET family, GSK1210151A (I-BET151), has profound efficacy against human and murine MLL-fusion leukaemic cell lines, through the induction of early cell cycle arrest and apoptosis. I-BET151 treatment in two human leukaemia cell lines with different MLL fusions alters the expression of a common set of genes whose function may account for these phenotypic changes. The mode of action of I-BET151 is, at least in part, due to the inhibition of transcription at key genes (BCL2, C-MYC and CDK6) through the displacement of BRD3/4, PAFc and SEC components from chromatin. In vivo studies indicate that I-BET151 has significant therapeutic value, providing survival benefit in two distinct mouse models of murine MLL-AF9 and human MLL-AF4 leukaemia. Finally, the efficacy of I-BET151 against human leukaemia stem cells is demonstrated, providing further evidence of its potent therapeutic potential. These findings establish the displacement of BET proteins from chromatin as a promising epigenetic therapy for these aggressive leukaemias.
