 网站首页
网站首页


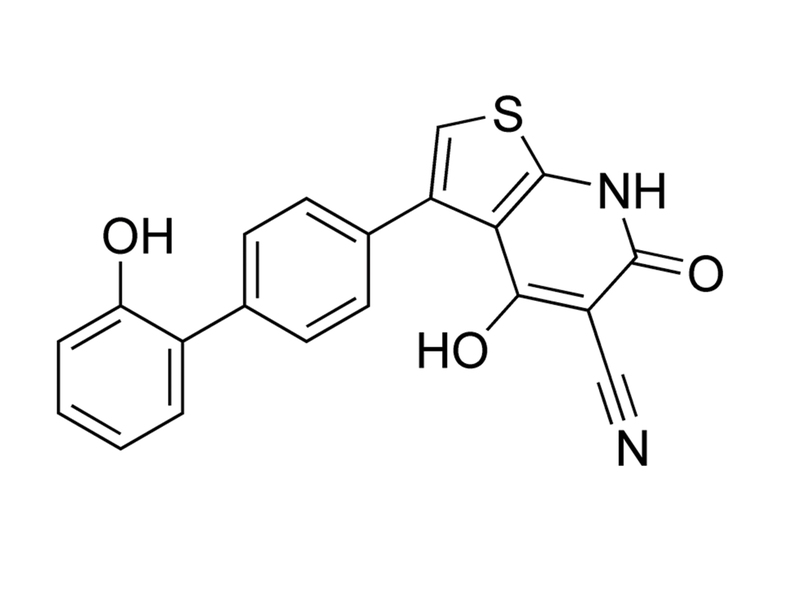
概要
A769662 is a cell-permeable, direct activator of AMP-activated protein kinase (AMPK) with an EC₅₀ of 116 nM (Goransson et al.). A 4.1-fold stimulation of AMPK is observed, via an allosteric mechanism, which potentially inhibits dephosphorylation on Thr172 (Goransson et al.; Sanders et al.). A769662 specifically activates β1 subunit–containing AMPK heterotrimers, and its effects are independent of kinases upstream of AMPK. Activation of AMPK can inhibit the mTORC1 signaling pathway (Huang et al.). A769662 is also an inhibitor of Na(+)-K(+)-ATPase (Benziane et al.).
REPROGRAMMING
· Inhibits reprogramming of mouse fibroblasts to induced pluripotent stem cells (Vazquez-Martin et al.).
MAINTENANCE
· Inhibits proliferation of mesenchymal stem cells (de Meester et al.).
CANCER RESEARCH
· Delays tumor onset in PTEN-deficient mice (Huang et al.).
METABOLISM
· Inhibits fatty acid synthesis in primary rat hepatocytes and lowers blood glucose in Sprague Dawley rats (Cool et al.).
REPROGRAMMING
· Inhibits reprogramming of mouse fibroblasts to induced pluripotent stem cells (Vazquez-Martin et al.).
MAINTENANCE
· Inhibits proliferation of mesenchymal stem cells (de Meester et al.).
CANCER RESEARCH
· Delays tumor onset in PTEN-deficient mice (Huang et al.).
METABOLISM
· Inhibits fatty acid synthesis in primary rat hepatocytes and lowers blood glucose in Sprague Dawley rats (Cool et al.).
技术资料
| Document Type | 产品名称 | Catalog # | Lot # | 语言 |
|---|---|---|---|---|
| Product Information Sheet | A769662 | 72922, 72924 | All | English |
| Safety Data Sheet | A769662 | 72922, 72924 | All | English |
数据及文献
Publications (7)
Cardiovascular research 2014
Role of AMP-activated protein kinase in regulating hypoxic survival and proliferation of mesenchymal stem cells.
Abstract
Abstract
AIMS: Mesenchymal stem cells (MSCs) are widely used for cell therapy, particularly for the treatment of ischaemic heart disease. Mechanisms underlying control of their metabolism and proliferation capacity, critical elements for their survival and differentiation, have not been fully characterized. AMP-activated protein kinase (AMPK) is a key regulator known to metabolically protect cardiomyocytes against ischaemic injuries and, more generally, to inhibit cell proliferation. We hypothesized that AMPK plays a role in control of MSC metabolism and proliferation. METHODS AND RESULTS: MSCs isolated from murine bone marrow exclusively expressed the AMPKα1 catalytic subunit. In contrast to cardiomyocytes, a chronic exposure of MSCs to hypoxia failed to induce cell death despite the absence of AMPK activation. This hypoxic tolerance was the consequence of a preference of MSC towards glycolytic metabolism independently of oxygen availability and AMPK signalling. On the other hand, A-769662, a well-characterized AMPK activator, was able to induce a robust and sustained AMPK activation. We showed that A-769662-induced AMPK activation inhibited MSC proliferation. Proliferation was not arrested in MSCs derived from AMPKα1-knockout mice, providing genetic evidence that AMPK is essential for this process. Among AMPK downstream targets proposed to regulate cell proliferation, we showed that neither the p70 ribosomal S6 protein kinase/eukaryotic elongation factor 2-dependent protein synthesis pathway nor p21 was involved, whereas p27 expression was increased by A-769662. Silencing p27 expression partially prevented the A-769662-dependent inhibition of MSC proliferation. CONCLUSION: MSCs resist hypoxia independently of AMPK whereas chronic AMPK activation inhibits MSC proliferation, p27 being involved in this regulation.
Cell cycle (Georgetown, Tex.) 2012 MAR
Activation of AMP-activated protein kinase (AMPK) provides a metabolic barrier to reprogramming somatic cells into stem cells.
Abstract
Abstract
The ability of somatic cells to reprogram their ATP-generating machinery into a Warburg-like glycolytic metabotype while overexpressing stemness genes facilitates their conversion into either induced pluripotent stem cells (iPSCs) or tumor-propagating cells. AMP-activated protein kinase (AMPK) is a metabolic master switch that senses and decodes intracellular changes in energy status; thus, we have evaluated the impact of AMPK activation in regulating the generation of iPSCs from nonstem cells of somatic origin. The indirect and direct activation of AMPK with the antidiabetic biguanide metformin and the thienopyridone A-769662, respectively, impeded the reprogramming of mouse embryonic and human diploid fibroblasts into iPSCs. The AMPK activators established a metabolic barrier to reprogramming that could not be bypassed, even through p53 deficiency, a fundamental mechanism to greatly improve the efficiency of stem-cell production. Treatment with metformin or A-769662 before the generation of iPSC colonies was sufficient to drastically decrease iPSC generation, suggesting that AMPK activation impedes early stem cell genetic reprogramming. Monitoring the transcriptional activation status of each individual reprogramming factor (i.e., Oct4, Sox2, Klf4 and c-Myc) revealed that AMPK activation notably prevented the transcriptional activation of Oct4, the master regulator of the pluripotent state. AMPK activation appears to impose a normalized metabolic flow away from the required pro-immortalizing glycolysis that fuels the induction of stemness and pluripotency, endowing somatic cells with an energetic infrastructure that is protected against reprogramming. AMPK-activating anti-reprogramming strategies may provide a roadmap for the generation of novel cancer therapies that metabolically target tumor-propagating cells.
American journal of physiology. Cell physiology 2009
AMP-activated protein kinase activator A-769662 is an inhibitor of the Na(+)-K(+)-ATPase.
Abstract
Abstract
Muscle contraction and metabolic stress are potent activators of AMP-activated protein kinase (AMPK). AMPK restores energy balance by activating processes that produce energy while inhibiting those that consume energy. The role of AMPK in the regulation of active ion transport is unclear. Our aim was to determine the effect of the AMPK activator A-769662 on Na(+)-K(+)-ATPase function in skeletal muscle cells. Short-term incubation of differentiated rat L6 myotubes with 100 microM A-769662 increased AMPK and acetyl-CoA carboxylase (ACC) phosphorylation in parallel with decreased Na(+)-K(+)-ATPase alpha(1)-subunit abundance at the plasma membrane and ouabain-sensitive (86)Rb(+) uptake. Notably, the effect of A-769662 on Na(+)-K(+)-ATPase was similar in muscle cells that do not express AMPK alpha(1)- and alpha(2)-catalytic subunits. A-769662 directly inhibits the alpha(1)-isoform of the Na(+)-K(+)-ATPase, purified from rat and human kidney cells in vitro with IC(50) 57 microM and 220 microM, respectively. Inhibition of the Na(+)-K(+)-ATPase by 100 microM ouabain decreases sodium pump activity and cell surface abundance, similar to the effect of A-769662, without affecting AMPK and ACC phosphorylation. In conclusion, the AMPK activator A-769662 inhibits Na(+)-K(+)-ATPase activity and decreases the sodium pump cell surface abundance in L6 skeletal muscle cells. The effect of A-769662 on sodium pump is due to direct inhibition of the Na(+)-K(+)-ATPase activity, rather than AMPK activation. This AMPK-independent effect on Na(+)-K(+)-ATPase calls into question the use of A-769662 as a specific AMPK activator for metabolic studies.
The Biochemical journal 2008
Important role of the LKB1-AMPK pathway in suppressing tumorigenesis in PTEN-deficient mice.
Abstract
Abstract
The LKB1 tumour suppressor phosphorylates and activates AMPK (AMP-activated protein kinase) when cellular energy levels are low, thereby suppressing growth through multiple pathways, including inhibiting the mTORC1 (mammalian target of rapamycin complex 1) kinase that is activated in the majority of human cancers. Blood glucose-lowering Type 2 diabetes drugs also induce LKB1 to activate AMPK, indicating that these compounds could be used to suppress growth of tumour cells. In the present study, we investigated the importance of the LKB1-AMPK pathway in regulating tumorigenesis in mice resulting from deficiency of the PTEN (phosphatase and tensin homologue deleted on chromosome 10) tumour suppressor, which drives cell growth through overactivation of the Akt and mTOR (mammalian target of rapamycin) kinases. We demonstrate that inhibition of AMPK resulting from a hypomorphic mutation that decreases LKB1 expression does not lead to tumorigenesis on its own, but markedly accelerates tumour development in PTEN(+/-) mice. In contrast, activating the AMPK pathway by administration of metformin, phenformin or A-769662 to PTEN(+/-) mice significantly delayed tumour onset. We demonstrate that LKB1 is required for activators of AMPK to inhibit mTORC1 signalling as well as cell growth in PTEN-deficient cells. Our findings highlight, using an animal model relevant to understanding human cancer, the vital role that the LKB1-AMPK pathway plays in suppressing tumorigenesis resulting from loss of the PTEN tumour suppressor. They also suggest that pharmacological inhibition of LKB1 and/or AMPK would be undesirable, at least for the treatment of cancers in which the mTORC1 pathway is activated. Most importantly, our results demonstrate the potential of AMPK activators, such as clinically approved metformin, as anticancer agents, which will suppress tumour development by triggering a physiological signalling pathway that potently inhibits cell growth.
The Journal of biological chemistry 2007
Defining the mechanism of activation of AMP-activated protein kinase by the small molecule A-769662, a member of the thienopyridone family.
Abstract
Abstract
AMP-activated protein kinase (AMPK) plays a key role in maintaining energy homeostasis. Activation of AMPK in peripheral tissues has been shown to alleviate the symptoms of metabolic diseases, such as type 2 diabetes, and consequently AMPK is a target for treatment of these diseases. Recently, a small molecule activator (A-769662) of AMPK was identified that had beneficial effects on metabolism in ob/ob mice. Here we show that A-769662 activates AMPK both allosterically and by inhibiting dephosphorylation of AMPK on Thr-172, similar to the effects of AMP. A-769662 activates AMPK harboring a mutation in the gamma subunit that abolishes activation by AMP. An AMPK complex lacking the glycogen binding domain of the beta subunit abolishes the allosteric effect of A-769662 but not the allosteric activation by AMP. Moreover, mutation of serine 108 to alanine, an autophosphorylation site within the glycogen binding domain of the beta1 subunit, almost completely abolishes activation of AMPK by A-769662 in cells and in vitro, while only partially reducing activation by AMP. Based on our results we propose a model for activation of AMPK by A-769662. Importantly, this model may provide clues for understanding the mechanism by which AMP leads to activation of AMPK, which in turn may help in the identification of other AMPK activators.
Journal of Biological Chemistry 2007
Mechanism of Action of A-769662, a Valuable Tool for Activation of AMP-activated Protein Kinase
Abstract
Abstract
We have studied the mechanism of A-769662, a new activator of AMP-activated protein kinase (AMPK). Unlike other pharmacological activators, it directly activates native rat AMPK by mimicking both effects of AMP, i.e. allosteric activation and inhibition of dephosphorylation. We found that it has no effect on the isolated alpha subunit kinase domain, with or without the associated autoinhibitory domain, or on interaction of glycogen with the beta subunit glycogen-binding domain. Although it mimics actions of AMP, it has no effect on binding of AMP to the isolated Bateman domains of the gamma subunit. The addition of A-769662 to mouse embryonic fibroblasts or primary mouse hepatocytes stimulates phosphorylation of acetyl-CoA carboxylase (ACC), effects that are completely abolished in AMPK-alpha1(-/-)alpha2(-/-) cells but not in TAK1(-/-) mouse embryonic fibroblasts. Phosphorylation of AMPK and ACC in response to A-769662 is also abolished in isolated mouse skeletal muscle lacking LKB1, a major upstream kinase for AMPK in this tissue. However, in HeLa cells, which lack LKB1 but express the alternate upstream kinase calmodulin-dependent protein kinase kinase-beta, phosphorylation of AMPK and ACC in response to A-769662 still occurs. These results show that in intact cells, the effects of A-769662 are independent of the upstream kinase utilized. We propose that this direct and specific AMPK activator will be a valuable experimental tool to understand the physiological roles of AMPK.
