 网站首页
网站首页


概要
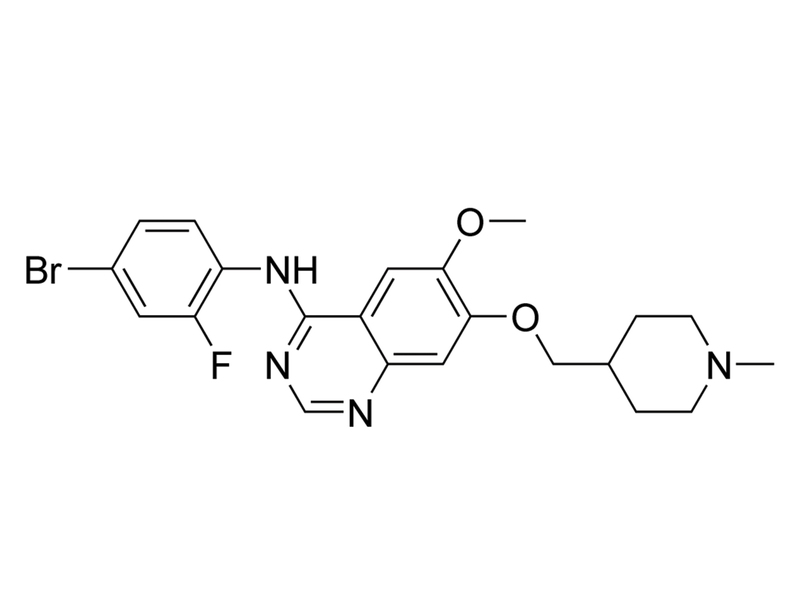
Vandetanib is a multi-kinase inhibitor targeting primarily receptor tyrosine kinases, such as vascular endothelial growth factor receptors (VEGFR1, KDR, and FLT4), epidermal growth factor receptor (EGFR), and fibroblast growth factor receptor (FGFR; Morabito et al.). It most potently inhibits KDR with an IC₅₀ of 40 nM. It also inhibits non-receptor tyrosine kinases such as RET, ABL, and SRC as well as several serine/threonine kinases (Carlomagno et al.; Hennequin et al.; Kiselyov et al.; Levinson & Boxer).
CANCER RESEARCH
· Inhibits tumor growth in mouse xenograft models, including RET/PTC papillary thyroid carcinoma and lung carcinoma (Hennequin et al.; Carlomagno et al.).
· Inhibits angiogenesis, cell growth, and metastasis in numerous cancer cell lines (Morabito et al.; Wedge et al.).
CANCER RESEARCH
· Inhibits tumor growth in mouse xenograft models, including RET/PTC papillary thyroid carcinoma and lung carcinoma (Hennequin et al.; Carlomagno et al.).
· Inhibits angiogenesis, cell growth, and metastasis in numerous cancer cell lines (Morabito et al.; Wedge et al.).
技术资料
| Document Type | 产品名称 | Catalog # | Lot # | 语言 |
|---|---|---|---|---|
| Product Information Sheet | Vandetanib | 73532, 73534 | All | English |
| Safety Data Sheet | Vandetanib | 73532, 73534 | All | English |
数据及文献
Publications (6)
PloS one 2012
Structural and spectroscopic analysis of the kinase inhibitor bosutinib and an isomer of bosutinib binding to the Abl tyrosine kinase domain.
Abstract
Abstract
Chronic myeloid leukemia (CML) is caused by the kinase activity of the BCR-Abl fusion protein. The Abl inhibitors imatinib, nilotinib and dasatinib are currently used to treat CML, but resistance to these inhibitors is a significant clinical problem. The kinase inhibitor bosutinib has shown efficacy in clinical trials for imatinib-resistant CML, but its binding mode is unknown. We present the 2.4 Å structure of bosutinib bound to the kinase domain of Abl, which explains the inhibitor's activity against several imatinib-resistant mutants, and reveals that similar inhibitors that lack a nitrile moiety could be effective against the common T315I mutant. We also report that two distinct chemical compounds are currently being sold under the name bosutinib"�
The oncologist 2009
Vandetanib (ZD6474), a dual inhibitor of vascular endothelial growth factor receptor (VEGFR) and epidermal growth factor receptor (EGFR) tyrosine kinases: current status and future directions.
Abstract
Abstract
Vandetanib is a novel, orally available inhibitor of different intracellular signaling pathways involved in tumor growth, progression, and angiogenesis: vascular endothelial growth factor receptor-2, epidermal growth factor receptor, and REarranged during Transfection tyrosine kinase activity. Phase I clinical trials have shown that vandetanib is well tolerated as a single agent at daily doses textless or =300 mg. In the phase II setting, negative results were observed with vandetanib in small cell lung cancer, metastatic breast cancer, and multiple myeloma. In contrast, three randomized phase II studies showed that vandetanib prolonged the progression-free survival (PFS) time of patients with non-small cell lung cancer (NSCLC) as a single agent when compared with gefitinib or when added to chemotherapy. Rash, diarrhea, hypertension, fatigue, and asymptomatic QTc prolongation were the most common adverse events. Antitumor activity was also observed in medullary thyroid cancer. Four randomized phase III clinical trials in NSCLC are exploring the efficacy of vandetanib in combination with docetaxel, the Zactima in cOmbination with Docetaxel In non-small cell lung Cancer (ZODIAC) trial, or with pemetrexed, the Zactima Efficacy with Alimta in Lung cancer (ZEAL) trial, or as a single agent, the Zactima Efficacy when Studied versus Tarceva (ZEST) and the Zactima Efficacy trial for NSCLC Patients with History of EGFR-TKI chemo-Resistance (ZEPHYR) trials. Based on a press release by the sponsor of these trials, the PFS time was longer with vandetanib in the ZODIAC and ZEAL trials; the ZEST trial was negative for its primary superiority analysis, but was successful according to a preplanned noninferiority analysis of PFS. Ongoing phase II and III clinical trials will better define the appropriate schedule, the optimal setting of evaluation, and the safety of long-term use of vandetanib.
Chemical biology & drug design 2007
1H-1,2,4-triazol-3-yl-anilines: novel potent inhibitors of vascular endothelial growth factor receptors 1 and 2.
Abstract
Abstract
Novel derivatives of 1,2,4-triazoles are described as potent ATP-competitive inhibitors of vascular endothelial growth factor receptors I and II (VEGFR-1/2). A number of compounds display VEGFR-2 inhibitory activity comparable to that of Vatalanib and Vandetanib in both homogenous time-resolved fluorescence enzymatic and cellular assays. Several active molecules feature high intrinsic permeability (textgreater30 x 10(-5) cm/min) across Caco-2 cell monolayer.
Cancer research 2002 DEC
ZD6474, an orally available inhibitor of KDR tyrosine kinase activity, efficiently blocks oncogenic RET kinases.
Abstract
Abstract
RET/papillary thyroid carcinoma (PTC) oncogenes, generated by recombination of the tyrosine kinase-encoding domain of RET with different heterologous genes, are prevalent in papillary carcinomas of the thyroid. Point mutations of RET cause multiple endocrine neoplasia type 2 (MEN2) familial cancer syndrome and are found in sporadic medullary thyroid carcinomas. Here, we show that ZD6474, a low molecular weight tyrosine kinase inhibitor, blocks the enzymatic activity of RET-derived oncoproteins at a one-half maximal inhibitory concentration of 100 nM. ZD6474 blocked in vivo phosphorylation and signaling of the RET/PTC3 and RET/MEN2B oncoproteins and of an epidermal growth factor (EGF)-activated EGF-receptor/RET chimeric receptor. RET/PTC3-transformed cells-treated ZD6474 lost proliferative autonomy and showed morphological reversion. ZD6474 prevented the growth of two human PTC cell lines that carry spontaneous RET/PTC1 rearrangements. Finally, it blocked anchorage-independent growth of RET/PTC3-transformed NIH3T3 fibroblasts and the formation of tumors after injection of NIH-RET/PTC3 cells into nude mice. Thus, targeting RET oncogenes with ZD6474 might offer a potential treatment strategy for carcinomas sustaining oncogenic activation of RET.
Journal of medicinal chemistry 2002
Novel 4-anilinoquinazolines with C-7 basic side chains: design and structure activity relationship of a series of potent, orally active, VEGF receptor tyrosine kinase inhibitors.
Abstract
Abstract
We have previously shown that 4-anilinoquinazolines can be potent inhibitors of vascular endothelial growth factor (VEGF) receptor (Flt-1 and KDR) tyrosine kinase activity. A novel subseries of 4-anilinoquinazolines that possess basic side chains at the C-7 position of the quinazoline nucleus have been synthesized. This subseries contains potent, nanomolar inhibitors of KDR (median IC(50) 0.02 microM, range 0.001-0.04 microM), which are comparatively less potent vs Flt-1 tyrosine kinase (median IC(50) 0.55 microM, range 0.02-1.6 microM). The compounds also retain some inhibitory activity against the tyrosine kinase associated to the endothelial growth factor receptor (EGFR) (median IC(50) 0.2 microM, range 0.075-0.8 microM) but demonstrate selectivity vs that associated to the FGF receptor 1 (median IC(50) 2.5 microM, range 0.9-19 microM). This selectivity profile is also evident in a growth factor-stimulated human endothelial cell (HUVEC) proliferation assay (i.e., inhibition of VEGF textgreater EGF textgreater FGF), with inhibition of VEGF-induced proliferation being achieved at nanomolar concentrations (median IC(50) 0.06 microM). Further examination of compound 2 (ZD6474) in recombinant enzyme assays revealed excellent selectivity for the inhibition of KDR tyrosine kinase (IC(50) 0.04 microM) vs the kinase activity of erbB2, MEK, CDK-2, Tie-2, IGFR-1R, PDK, PDGFRbeta, and AKT (IC(50) range: 1.1 to textgreater100 microM). Anilinoquinazolines possessing basic C-7 side chains exhibited markedly improved aqueous solubility over previously described anilinoquinazolines possessing neutral C-7 side chains (up to 500-fold improvement at pH 7.4). In addition, aqueous solubility of the neutral fraction present at pH 7.4 of the basic subseries of anilinoquinazoline proved to be higher than that of the neutral analogue 1 (ZD4190). Oral administration of representative compounds to mice (50 mg/kg) produced plasma levels between 0.2 and 3 microM at 24 h after dosing. Our development candidate 2 demonstrated a very attractive in vitro profile combined with excellent solubility (330 microM at pH 7.4) and good oral bioavailability in rat and dog (textgreater 80 and textgreater 50%, respectively). This compound demonstrated highly significant, dose-dependent, antitumor activity in athymic mice. Once daily oral administration of 100 mg/kg of compound 2 for 21 days inhibited the growth of established Calu-6 lung carcinoma xenografts by 79% (P textless 0.001, Mann Whitney rank sum test), and substantial inhibition (36%, P textless 0.02) was evident with 12.5 mg/kg/day.
Cancer research 2002
ZD6474 inhibits vascular endothelial growth factor signaling, angiogenesis, and tumor growth following oral administration.
Abstract
Abstract
ZD6474 [N-(4-bromo-2-fluorophenyl)-6-methoxy-7-[(1-methylpiperidin-4-yl)methoxy]quinazolin-4-amine]is a potent, p.o. active, low molecular weight inhibitor of kinase insert domain-containing receptor [KDR/vascular endothelial growth factor receptor (VEGFR) 2] tyrosine kinase activity (IC(50) = 40 nM). This compound has some additional activity versus the tyrosine kinase activity of fms-like tyrosine kinase 4 (VEGFR3;IC(50) = 110 nM) and epidermal growth factor receptor (EGFR/HER1; IC(50) = 500 nM) and yet demonstrates selectivity against a range of other tyrosine and serine-threonine kinases. The activity of ZD6474 versus KDR tyrosine kinase translates into potent inhibition of vascular endothelial growth factor-A (VEGF)-stimulated endothelial cell (human umbilical vein endothelial cell) proliferation in vitro (IC(50) = 60 nM). Selective inhibition of VEGF signaling has been demonstrated in vivo in a growth factor-induced hypotension model in anesthetized rat: administration of ZD6474 (2.5 mg/kg, i.v.) reversed a hypotensive change induced by VEGF (by 63%) but did not significantly affect that induced by basic fibroblast growth factor. Once-daily oral administration of ZD6474 to growing rats for 14 days produced a dose-dependent increase in the femoro-tibial epiphyseal growth plate zone of hypertrophy, which is consistent with inhibition of VEGF signaling and angiogenesis in vivo. Administration of 50 mg/kg/day ZD6474 (once-daily, p.o.) to athymic mice with intradermally implanted A549 tumor cells also inhibited tumor-induced neovascularization significantly (63% inhibition after 5 days; P textless 0.001). Oral administration of ZD6474 to athymic mice bearing established (0.15-0.47 cm(3)), histologically distinct (lung, prostate, breast, ovarian, colon, or vulval) human tumor xenografts or after implantation of aggressive syngeneic rodent tumors (lung, melanoma) in immunocompetent mice, produced a dose-dependent inhibition of tumor growth in all cases. Statistically significant antitumor activity was evident in each model with at least 25 mg/kg ZD6474 once daily (P textless 0.05, one-tailed t test). Histological analysis of Calu-6 tumors treated with 50 mg/kg/day ZD6474 for 24 days showed a significant reduction (textgreater70%) in CD31 (endothelial cell) staining in nonnecrotic regions. ZD6474 also restrained growth of much larger (0.9 cm(3) volume) Calu-6 lung tumor xenografts and induced profound regression in established PC-3 prostate tumors of 1.4 cm(3) volume. ZD6474 is currently in Phase I clinical development as a once-daily oral therapy in patients with advanced cancer.
