 网站首页
网站首页


概要
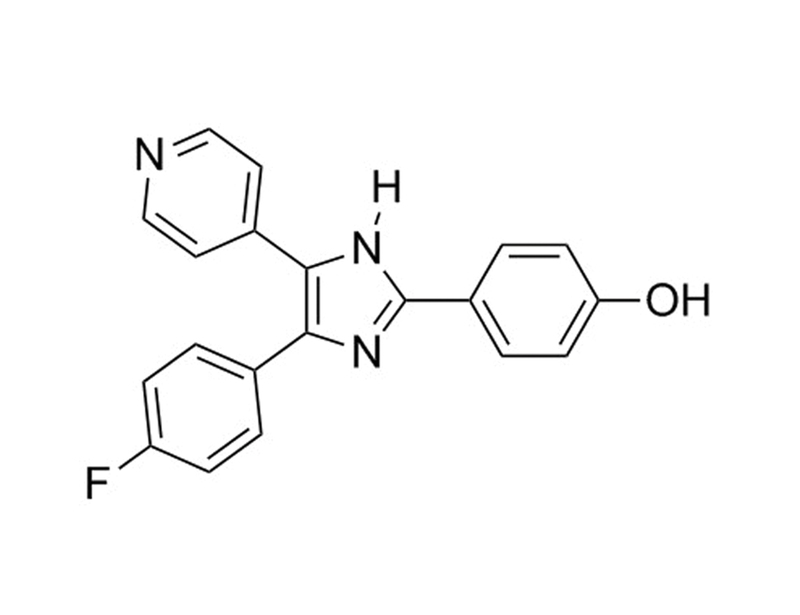
SB202190 is a selective, potent, cell-permeable inhibitor of p38 MAP kinases, inhibiting p38α (SAPK2A, MAPK14) and p38β (SAPK2B, MAPK11) with IC₅₀ values of 50 and 100 nM, respectively (Davies et al.; Jiang et al.). As a pyridinyl imidazole inhibitor, SB202190 directly binds p38 MAP kinases in the ATP binding pocket (Fox et al.).
MAINTENANCE AND SELF-RENEWAL
· Improves the self-renewal ability of neural stem cells from NPC1-deficient mice (Yang et al.).
· Blocks adiponectin-mediated proliferation of hematopoietic stem cells (DiMascio et al.).
· Reduces BMP3-mediated proliferation of C3H10T1/2 mesenchymal stem cells (Stewart et al.).
DIFFERENTIATION
· Induces cardiomyocyte differentiation from human embryonic stem cells (Graichen et al.).
MAINTENANCE AND SELF-RENEWAL
· Improves the self-renewal ability of neural stem cells from NPC1-deficient mice (Yang et al.).
· Blocks adiponectin-mediated proliferation of hematopoietic stem cells (DiMascio et al.).
· Reduces BMP3-mediated proliferation of C3H10T1/2 mesenchymal stem cells (Stewart et al.).
DIFFERENTIATION
· Induces cardiomyocyte differentiation from human embryonic stem cells (Graichen et al.).
技术资料
| Document Type | 产品名称 | Catalog # | Lot # | 语言 |
|---|---|---|---|---|
| Product Information Sheet | SB202190 | 72632, 72634 | All | English |
| Safety Data Sheet | SB202190 | 72632, 72634 | All | English |
数据及文献
Publications (7)
Journal of cellular physiology 2010 JUN
BMP-3 promotes mesenchymal stem cell proliferation through the TGF-beta/activin signaling pathway.
Abstract
Abstract
Adipogenesis plays a key role in the pathogenesis of obesity. It begins with the commitment of mesenchymal stem cells (MSCs) to the adipocyte lineage, followed by terminal differentiation of preadipocytes to mature adipocytes. A critical, but poorly understood, component of adipogenesis involves proliferation of MSCs and preadipocytes. The present study was undertaken to examine the hypothesis that bone morphogenetic protein-3 (BMP-3) promotes adipogenesis using C3H10T1/2 MSCs and 3T3-L1 preadipocytes as in vitro model systems. We demonstrated that although it did not promote the commitment of MSCs to the adipocyte lineage or the differentiation of preadipocytes to adipocytes, BMP-3-stimulated proliferation by threefold in both cell types. Owing to a lack of information on MSC proliferation, we then delineated the molecular mechanisms underlying BMP-3-stimulated MSC proliferation. We showed that BMP-3 activated the transforming growth factor-beta (TGF-beta)/activin but not ERK1/2, p38 MAPK, or JNK signaling pathways in C3H10T1/2 cells. Furthermore, the TGF-beta/activin receptor kinase inhibitor SB-431542 blocked BMP-3-stimulated proliferation. Importantly, siRNA-mediated knockdown of the key TGF-beta/activin signaling pathway components, ActRIIB, ALK4, or Smad2, abrogated the mitogenic effects of BMP-3 on MSCs. Together, these results demonstrate that BMP-3 stimulates MSC proliferation via the TGF-beta/activin signaling pathway, thus revealing a novel role for this divergent and poorly understood member of the TGF-beta superfamily in regulating MSC proliferation.
Differentiation 2008 APR
Enhanced cardiomyogenesis of human embryonic stem cells by a small molecular inhibitor of p38 MAPK.
Abstract
Abstract
Human embryonic stem cells (hESC) can differentiate to cardiomyocytes in vitro but with generally poor efficiency. Here, we describe a novel method for the efficient generation of cardiomyocytes from hESC in a scalable suspension culture process. Differentiation in serum-free medium conditioned by the cell line END2 (END2-CM) readily resulted in differentiated cell populations with more than 10% cardiomyocytes without further enrichment. By screening candidate molecules, we have identified SB203580, a specific p38 MAP kinase inhibitor, as a potent promoter of hESC-cardiogenesis. SB203580 at concentrations textless10 microM, induced more than 20% of differentiated cells to become cardiomyocytes and increased total cell numbers, so that the overall cardiomyocyte yield was approximately 2.5-fold higher than controls. Gene expression indicated that early mesoderm formation was favored in the presence of SB203580. Accordingly, transient addition of the inhibitor at the onset of differentiation only was sufficient to determine the hESC fate. Patch clamp electrophysiology showed that the distribution of cardiomyocyte phenotypes in the population was unchanged by the compound. Interestingly, cardiomyogenesis was strongly inhibited at SB203580 concentrations textgreater or =15 microM. Thus, modulation of the p38MAP kinase pathway, in combination with factors released by END2 cells, plays an essential role in early lineage determination in hESC and the efficiency of cardiomyogenesis. Our findings contribute to transforming human cardiomyocyte generation from hESC into a robust and scalable process.
The Journal of Immunology 2007 MAR
Identification of Adiponectin as a Novel Hemopoietic Stem Cell Growth Factor
Abstract
Abstract
The hemopoietic microenvironment consists of a diverse repertoire of cells capable of providing signals that influence hemopoietic stem cell function. Although the role of osteoblasts and vascular endothelial cells has recently been characterized, the function of the most abundant cell type in the bone marrow, the adipocyte, is less defined. Given the emergence of a growing number of adipokines, it is possible that these factors may also play a role in regulating hematopoiesis. Here, we investigated the role of adiponectin, a secreted molecule derived from adipocytes, in hemopoietic stem cell (HSC) function. We show that adiponectin is expressed by components of the HSC niche and its receptors AdipoR1 and AdipoR2 are expressed by HSCs. At a functional level, adiponectin influences HSCs by increasing their proliferation, while retaining the cells in a functionally immature state as determined by in vitro and in vivo assays. We also demonstrate that adiponectin signaling is required for optimal HSC proliferation both in vitro and in long term hemopoietic reconstitution in vivo. Finally we show that adiponectin stimulation activates p38 MAPK, and that inhibition of this pathway abrogates adiponectin's proliferative effect on HSCs. These studies collectively identify adiponectin as a novel regulator of HSC function and suggest that it acts through a p38 dependent pathway.
Stem cells 2006 FEB
NPC1 gene deficiency leads to lack of neural stem cell self-renewal and abnormal differentiation through activation of p38 mitogen-activated protein kinase signaling.
Abstract
Abstract
Neural stem cells (NSCs) are capable of giving rise to neurons, glia, and astrocytes. Although self-renewal and differentiation in NSCs are regulated by many genes, such as Notch and Numb, little is known about the role of defective genes on the self-renewal and differentiation of NSCs from developing brain. The Niemann-Pick type C1 (NPC1) disease is a neurodegenerative disease caused by a mutation of the NPC1 gene that affects the function of the NPC1 protein. The ability of NSC self-renewal and differentiation was investigated using a model of NPC1 disease. The NPC1 disorder significantly affected the self-renewal ability of NSCs, as well as the differentiation. NSCs from NPC1-/- mice showed impaired self-renewal ability compared with the NPC1+/+ mice. These alterations were accompanied by the enhanced activity of p38 mitogen-activated protein kinases (MAPKs). Further, the specific p38 MAPK inhibitor SB202190 improved the self-renewal ability of NSCs from NPC-/- mice. This indicated that the NPC1 deficiency can lead to lack of self-renewal and altered differentiation of NSCs mediated by the activation of p38 MAPK, impairing the generation of neurospheres from NPC1-/- Thus, the NPC1 gene may play a crucial role in NSC self-renewal associated with p38 MAPK.
The Biochemical journal 2000 OCT
Specificity and mechanism of action of some commonly used protein kinase inhibitors.
Abstract
Abstract
The specificities of 28 commercially available compounds reported to be relatively selective inhibitors of particular serine/threonine-specific protein kinases have been examined against a large panel of protein kinases. The compounds KT 5720, Rottlerin and quercetin were found to inhibit many protein kinases, sometimes much more potently than their presumed targets, and conclusions drawn from their use in cell-based experiments are likely to be erroneous. Ro 318220 and related bisindoylmaleimides, as well as H89, HA1077 and Y 27632, were more selective inhibitors, but still inhibited two or more protein kinases with similar potency. LY 294002 was found to inhibit casein kinase-2 with similar potency to phosphoinositide (phosphatidylinositol) 3-kinase. The compounds with the most impressive selectivity profiles were KN62, PD 98059, U0126, PD 184352, rapamycin, wortmannin, SB 203580 and SB 202190. U0126 and PD 184352, like PD 98059, were found to block the mitogen-activated protein kinase (MAPK) cascade in cell-based assays by preventing the activation of MAPK kinase (MKK1), and not by inhibiting MKK1 activity directly. Apart from rapamycin and PD 184352, even the most selective inhibitors affected at least one additional protein kinase. Our results demonstrate that the specificities of protein kinase inhibitors cannot be assessed simply by studying their effect on kinases that are closely related in primary structure. We propose guidelines for the use of protein kinase inhibitors in cell-based assays.
Protein science 1998 NOV
A single amino acid substitution makes ERK2 susceptible to pyridinyl imidazole inhibitors of p38 MAP kinase.
Abstract
Abstract
Mitogen-activated protein (MAP) kinases are serine/threonine kinases that mediate intracellular signal transduction pathways. Pyridinyl imidazole compounds block pro-inflammatory cytokine production and are specific p38 kinase inhibitors. ERK2 is related to p38 in sequence and structure, but is not inhibited by pyridinyl imidazole inhibitors. Crystal structures of two pyridinyl imidazoles complexed with p38 revealed these compounds bind in the ATP site. Mutagenesis data suggested a single residue difference at threonine 106 between p38 and other MAP kinases is sufficient to confer selectivity of pyridinyl imidazoles. We have changed the equivalent residue in human ERK2, Q105, into threonine and alanine, and substituted four additional ATP binding site residues. The single residue change Q105A in ERK2 enhances the binding of SB202190 at least 25,000-fold compared to wild-type ERK2. We report enzymatic analyses of wild-type ERK2 and the mutant proteins, and the crystal structure of a pyridinyl imidazole, SB203580, bound to an ERK2 pentamutant, I103L, Q105T, D106H, E109G. T110A. These ATP binding site substitutions induce low nanomolar sensitivity to pyridinyl imidazoles. Furthermore, we identified 5-iodotubercidin as a potent ERK2 inhibitor, which may help reveal the role of ERK2 in cell proliferation.
