 网站首页
网站首页

概要
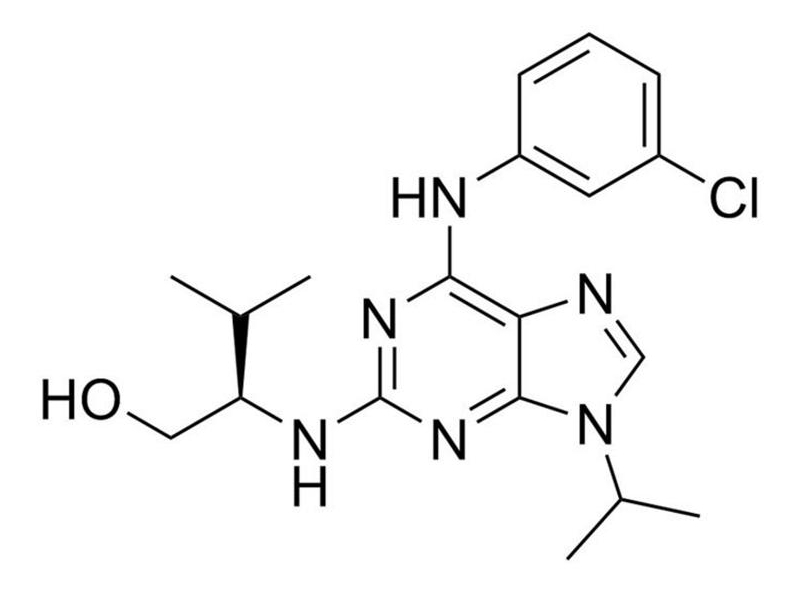
Purvalanol A is a cell-permeable, potent, and selective inhibitor of cyclin-dependent kinases (CDKs). CDKs and cyclins form a stoichiometric complex which is necessary for the CDK subunit to gain its protein kinase activity. It has been shown that these CDK/cyclin complexes play a key role in initiating G2/M transitions of the cell cycle (Jackman & Pines). Purvalanol A acts through competitive inhibition of ATP binding, to inhibit CDK1/cyclin B (IC₅₀ = 4 nM), CDK2/cyclin A (IC₅₀ = 70 nM), CDK2/cyclin E (IC₅₀ = 35 nM), CDK4/cyclin D1 (IC₅₀ = 850 nM) and CDK5-p35 (IC₅₀ = 75 nM; Bain et al.; Gray et al.), thereby arresting cells in G1 and G2.
CANCER RESEARCH
· Inhibits proliferation in exponentially growing cancer cell lines and reversibly arrests synchronised cells in G1 and G2 phase of cell cycle (Villerbu et al.).
· Induces apoptosis in MCF-7 estrogen receptor positive breast cancer cells (Obakan et al.).
· Suppresses cancer progression associated with Src up-regulation by the coordinated inhibition of cell cycle progression and tyrosine kinase signaling (Hikita et al.).
CANCER RESEARCH
· Inhibits proliferation in exponentially growing cancer cell lines and reversibly arrests synchronised cells in G1 and G2 phase of cell cycle (Villerbu et al.).
· Induces apoptosis in MCF-7 estrogen receptor positive breast cancer cells (Obakan et al.).
· Suppresses cancer progression associated with Src up-regulation by the coordinated inhibition of cell cycle progression and tyrosine kinase signaling (Hikita et al.).
技术资料
| Document Type | 产品名称 | Catalog # | Lot # | 语言 |
|---|---|---|---|---|
| Product Information Sheet | Purvalanol A | 73774 | All | English |
| Safety Data Sheet | Purvalanol A | 73774 | All | English |
数据及文献
Publications (6)
Molecular biology reports 2014 JAN
Purvalanol A is a strong apoptotic inducer via activating polyamine catabolic pathway in MCF-7 estrogen receptor positive breast cancer cells.
Abstract
Abstract
Purvalanol A is a specific CDK inhibitor which triggers apoptosis by causing cell cycle arrest in cancer cells. Although it has strong apoptotic potential, the mechanistic action of Purvalanol A on significant cell signaling targets has not been clarified yet. Polyamines are crucial metabolic regulators affected by CDK inhibition because of their role in cell cycle progress as well. In addition, malignant cells possess impaired polyamine homeostasis with high level of intracellular polyamines. Especially induction of polyamine catabolic enzymes spermidine/spermine N1-acetyltransferase (SSAT), polyamine oxidase (PAO) and spermine oxidase (SMO) induced toxic by-products in correlation with the induction of apoptosis in cancer cells. In this study, we showed that Purvalanol A induced apoptosis in caspase- dependent manner in MCF-7 ER(+) cells, while MDA-MB-231 (ER-) cells were less sensitive against drug. In addition Bcl-2 is a critical target for Purvalanol A, since Bcl-2 overexpressed cells are more resistant to Purvalanol A-mediated apoptosis. Furthermore, exposure of MCF-7 cells to Purvalanol A triggered SSAT and PAO upregulation and the presence of PAO/SMO inhibitor, MDL 72,527 prevented Purvalanol A-induced apoptosis.
Genes to cells : devoted to molecular & cellular mechanisms 2010 OCT
Purvalanol A, a CDK inhibitor, effectively suppresses Src-mediated transformation by inhibiting both CDKs and c-Src.
Abstract
Abstract
The nonreceptor tyrosine kinase c-Src is frequently over-expressed or hyperactivated in various human cancers and contributes to cancer progression in cooperation with up-regulated growth factor receptors. However, Src-selective anticancer drugs are still in clinical trials. To identify more effective inhibitors of c-Src-mediated cancer progression, we developed a new screening platform using Csk-deficient cells that can be transformed by c-Src. We found that purvalanol A, developed as a CDK inhibitor, potently suppressed the anchorage-independent growth of c-Src-transformed cells, indicating that the activation of CDKs contributes to the c-Src transformation. We also found that purvalanol A suppressed the c-Src activity as effectively as the Src-selective inhibitor PP2, and that it reverted the transformed morphology to a nearly normal shape with less cytotoxicity than PP2. Purvalanol A induced a strong G2-M arrest, whereas PP2 weakly acted on the G1-S transition. Furthermore, when compared with PP2, purvalanol A more effectively suppressed the growth of human colon cancer HT29 and SW480 cells, in which Src family kinases and CDKs are activated. These findings demonstrate that the coordinated inhibition of cell cycle progression and tyrosine kinase signaling by the multi-selective purvalanol A is effective in suppressing cancer progression associated with c-Src up-regulation.
The Biochemical journal 2003 APR
The specificities of protein kinase inhibitors: an update.
Abstract
Abstract
We have previously examined the specificities of 28 commercially available compounds, reported to be relatively selective inhibitors of particular serine/threonine-specific protein kinases [Davies, Reddy, Caivano and Cohen (2000) Biochem. J. 351, 95-105]. In the present study, we have extended this analysis to a further 14 compounds. Of these, indirubin-3'-monoxime, SP 600125, KT 5823 and ML-9 were found to inhibit a number of protein kinases and conclusions drawn from their use in cell-based assays are likely to be erroneous. Kenpaullone, Alsterpaullone, Purvalanol, Roscovitine, pyrazolopyrimidine 1 (PP1), PP2 and ML-7 were more specific, but still inhibited two or more protein kinases with similar potency. Our results suggest that the combined use of Roscovitine and Kenpaullone may be useful for identifying substrates and physiological roles of cyclin-dependent protein kinases, whereas the combined use of Kenpaullone and LiCl may be useful for identifying substrates and physiological roles of glycogen synthase kinase 3. The combined use of SU 6656 and either PP1 or PP2 may be useful for identifying substrates of Src family members. Epigallocatechin 3-gallate, one of the main polyphenolic constituents of tea, inhibited two of the 28 protein kinases in the panel, dual-specificity, tyrosine-phosphorylated and regulated kinase 1A (DYRK1A; IC(50)=0.33 microM) and p38-regulated/activated kinase (PRAK; IC(50)=1.0 microM).
International journal of cancer 2002 FEB
Cellular effects of purvalanol A: a specific inhibitor of cyclin-dependent kinase activities.
Abstract
Abstract
We have studied the effects of purvalanol A on the cell cycle progression, proliferation and viability. In synchronized cells, purvalanol A induced a reversible arrest the progression in G1 and G2 phase of the cell cycle, but did not prevent the completion of DNA synthesis in S-phase cells. The specificity of action of the drug was supported by the selective inhibition of the phosphorylation of cyclin-dependent kinase (cdk) substrates such as Rb and cyclin E. The cell contents of cyclins D1 and E were lower in cells incubated with purvalanol A compared to controls, but the level of the cdk inhibitory protein p21(WAF1/CIP1) was increased, indicating that the drug did not cause a general inhibition of gene expression. Purvalanol A did not inhibit transcription under cell-free conditions. This compound, however, caused an inhibition of the estradiol-induced expression of an integrated luciferase gene, suggesting that cdk or related enzymes may participate in the regulation of the activity of certain promoters. When exponentially growing cells, both mouse fibroblasts and human cancer cell lines, were incubated with purvalanol A for prolonged periods of time (24 hr), a lasting inhibition of cell proliferation as well as cell death were observed. In contrast, a 24 hr incubation of quiescent (non-transformed) cells with purvalanol A did not prevent their resumption of cell cycle after removal of the drug.
Science (New York, N.Y.) 1998 JUL
Exploiting chemical libraries, structure, and genomics in the search for kinase inhibitors.
Abstract
Abstract
Selective protein kinase inhibitors were developed on the basis of the unexpected binding mode of 2,6,9-trisubstituted purines to the adenosine triphosphate-binding site of the human cyclin-dependent kinase 2 (CDK2). By iterating chemical library synthesis and biological screening, potent inhibitors of the human CDK2-cyclin A kinase complex and of Saccharomyces cerevisiae Cdc28p were identified. The structural basis for the binding affinity and selectivity was determined by analysis of a three-dimensional crystal structure of a CDK2-inhibitor complex. The cellular effects of these compounds were characterized in mammalian cells and yeast. In the latter case the effects were characterized on a genome-wide scale by monitoring changes in messenger RNA levels in treated cells with high-density oligonucleotide probe arrays. Purine libraries could provide useful tools for analyzing a variety of signaling and regulatory pathways and may lead to the development of new therapeutics.
Cancer surveys 1997 JAN
Cyclins and the G2/M transition.
Abstract
Abstract
The entry of a cell into mitosis is regulated by an elaborate network of kinases and phosphatases that control both for the timing of cell division and the complete reorganization of the cellular architecture. The mitotic cyclin/Cdks form part of large multiprotein complexes whose other components are only now beginning to be identified. The continuing identification of proteins that contribute to these complexes and changes in the composition of these complexes are likely to give a more integrated view of how mitotic cyclin/Cdk complexes are regulated and how they function-not only to induce mitosis, but also to aid further mitotic progression. Furthermore, assigning specific G2/M functions to distinct mitotic cyclin/Cdk complexes will require the identification of differences in substrate specificities between the mitotic cyclin/Cdk complexes, perhaps in parallel with specific cyclin knockouts in mice. Such investigations will be complicated by potential functional overlap between mitotic cyclin/Cdk complexes in vitro and in vivo. Although cyclin/Cdk1 is thought to be the major kinase that initiates the onset of mitosis, a more complete understanding of how cells move from G2 to a mitotic state will require further identification of kinases operating upstream, downstream and in parallel with Cdk1, their substrates and their relationship with one another during the G2/M transition.
