 网站首页
网站首页
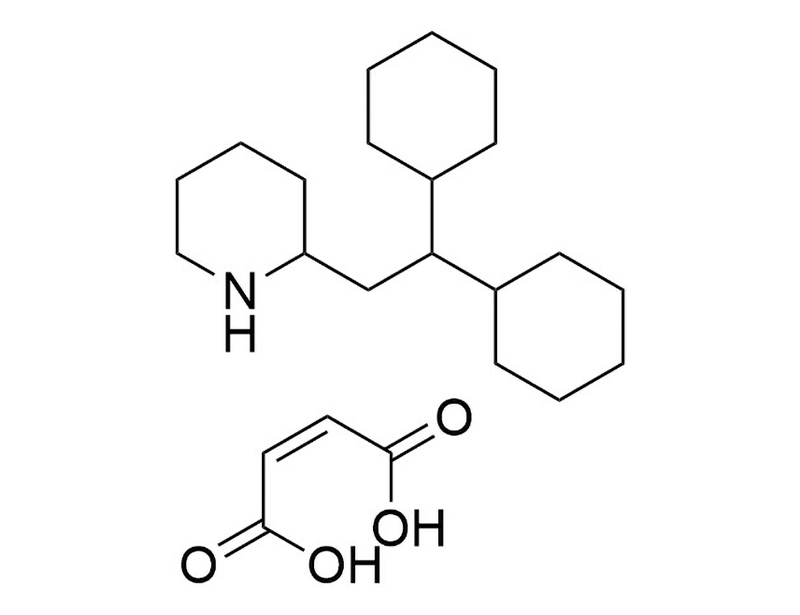


概要
Perhexiline Maleate is an inhibitor of the mitochondrial enzymes carnitine palmitoyltransferase 1 (CPT1; IC₅₀ = 0.077 mM in rat heart; IC₅₀ = 0.148 mM in rat liver) and CPT2 (IC₅₀ = 0.079 mM in rat heart; Kennedy et al. 1996; Kennedy et al. 2000). Perhexiline Maleate also modulates autophagy via mammalian target of rapamycin complex 1 (mTORC1) signaling (Balgi et al.).
METABOLISM
· Alters myocardial metabolism from fatty acid to glucose utilization, resulting in higher ATP production and oxygen consumption (Ashrafian et al.; Jeffrey et al.).
CANCER RESEARCH
· Inhibits mTORC1 signaling and induces autophagy in MCF-7 cells (Balgi et al.).
METABOLISM
· Alters myocardial metabolism from fatty acid to glucose utilization, resulting in higher ATP production and oxygen consumption (Ashrafian et al.; Jeffrey et al.).
CANCER RESEARCH
· Inhibits mTORC1 signaling and induces autophagy in MCF-7 cells (Balgi et al.).
技术资料
| Document Type | 产品名称 | Catalog # | Lot # | 语言 |
|---|---|---|---|---|
| Product Information Sheet | Perhexiline Maleate | 100-0267, 100-0268 | All | English |
| Safety Data Sheet | Perhexiline Maleate | 100-0267, 100-0268 | All | English |
数据及文献
Publications (5)
PloS one 2009 sep
Screen for chemical modulators of autophagy reveals novel therapeutic inhibitors of mTORC1 signaling.
Abstract
Abstract
BACKGROUND Mammalian target of rapamycin complex 1 (mTORC1) is a protein kinase that relays nutrient availability signals to control numerous cellular functions including autophagy, a process of cellular self-eating activated by nutrient depletion. Addressing the therapeutic potential of modulating mTORC1 signaling and autophagy in human disease requires active chemicals with pharmacologically desirable properties. METHODOLOGY/PRINCIPAL FINDINGS Using an automated cell-based assay, we screened a collection of {\textgreater}3,500 chemicals and identified three approved drugs (perhexiline, niclosamide, amiodarone) and one pharmacological reagent (rottlerin) capable of rapidly increasing autophagosome content. Biochemical assays showed that the four compounds stimulate autophagy and inhibit mTORC1 signaling in cells maintained in nutrient-rich conditions. The compounds did not inhibit mTORC2, which also contains mTOR as a catalytic subunit, suggesting that they do not inhibit mTOR catalytic activity but rather inhibit signaling to mTORC1. mTORC1 inhibition and autophagosome accumulation induced by perhexiline, niclosamide or rottlerin were rapidly reversed upon drug withdrawal whereas amiodarone inhibited mTORC1 essentially irreversibly. TSC2, a negative regulator of mTORC1, was required for inhibition of mTORC1 signaling by rottlerin but not for mTORC1 inhibition by perhexiline, niclosamide and amiodarone. Transient exposure of immortalized mouse embryo fibroblasts to these drugs was not toxic in nutrient-rich conditions but led to rapid cell death by apoptosis in starvation conditions, by a mechanism determined in large part by the tuberous sclerosis complex protein TSC2, an upstream regulator of mTORC1. By contrast, transient exposure to the mTORC1 inhibitor rapamycin caused essentially irreversible mTORC1 inhibition, sustained inhibition of cell growth and no selective cell killing in starvation. CONCLUSION/SIGNIFICANCE The observation that drugs already approved for human use can reversibly inhibit mTORC1 and stimulate autophagy should greatly facilitate the preclinical and clinical testing of mTORC1 inhibition for indications such as tuberous sclerosis, diabetes, cardiovascular disease and cancer.
Cardiovascular drug reviews 2007
Perhexiline.
Abstract
Abstract
Perhexiline, 2-(2,2-dicyclohexylethyl)piperidine, was originally developed as an anti-anginal drug in the 1970s. Despite its success, its use diminished due to the occurrence of poorly understood side effects including neurotoxicity and hepatotoxicity in a small proportion of patients. Recently, perhexiline's mechanism of action and the molecular basis of its toxicity have been elucidated. Perhexiline reduces fatty acid metabolism through the inhibition of carnitine palmitoyltransferase, the enzyme responsible for mitochondrial uptake of long-chain fatty acids. The corresponding shift to greater carbohydrate utilization increases myocardial efficiency (work done per unit oxygen consumption) and this oxygen-sparing effect explains its antianginal efficacy. Perhexiline's side effects are attributable to high plasma concentrations occurring with standard doses in patients with impaired metabolism due to CYP2D6 mutations. Accordingly, dose modification in these poorly metabolizing patients identified through therapeutic plasma monitoring can eliminate any significant side effects. Herein we detail perhexiline's pharmacology with particular emphasis on its mechanism of action and its side effects. We discuss how therapeutic plasma monitoring has led to perhexiline's safe reintroduction into clinical practice and how recent clinical data attesting to its safety and remarkable efficacy led to a renaissance in its use in both refractory angina and chronic heart failure. Finally, we discuss the application of pharmacogenetics in combination with therapeutic plasma monitoring to potentially broaden perhexiline's use in heart failure, aortic stenosis, and other cardiac conditions.
Journal of cardiovascular pharmacology 2000 dec
Effect of perhexiline and oxfenicine on myocardial function and metabolism during low-flow ischemia/reperfusion in the isolated rat heart.
Abstract
Abstract
Perhexiline is a potent prophylactic anti-anginal agent that has been shown to inhibit myocardial utilization of long-chain fatty acids and to inhibit the mitochondrial enzyme carnitine palmitoyltransferase (CPT)-1. We compared the hemodynamic and biochemical effects of perhexiline (0.5 and 2.0 microM) and of another CPT-1 inhibitor, oxfenicine (0.5 mM), in Langendorff-perfused rat hearts subjected to 60 min of low-flow ischemia (95{\%} flow reduction) followed by 30 min of reperfusion. Both perhexiline (2 microM only) and oxfenicine attenuated (p {\textless} 0.003, p {\textless} 0.0002, respectively) increases in diastolic tension during ischemia, without significant effects on developed tension, or on cardiac function during reperfusion. Myocardial concentrations of long-chain acylcarnitines (LCAC), products of CPT-1 action, were decreased (p {\textless} 0.05) by oxfenicine, unaffected by 2 microM perhexiline, and increased slightly by 0.5 microM perhexiline. Perhexiline, but not the active metabolite of oxfenicine, also inhibited cardiac CPT-2 with similar IC50 and Emax, although lower Hill slope, compared with CPT-1. Oxfenicine, but not perhexiline, reduced concentrations of the endogenous CPT-1 inhibitor, malonyl-CoA. Perhexiline, but not oxfenicine, inhibited myocardial release of lactate during normal flow. We conclude that (a) perhexiline protects against diastolic dysfunction during ischemia in this model, independent of major changes in LCAC accumulation and (b) this may result from simultaneous effects of perhexiline on myocardial CPT-1 and CPT-2.
Biochemical pharmacology 1996 jul
Inhibition of carnitine palmitoyltransferase-1 in rat heart and liver by perhexiline and amiodarone.
Abstract
Abstract
The mechanism of the anti-anginal effect of perhexiline is unclear but appears to involve a shift in cardiac metabolism from utilization of fatty acid to that of carbohydrate. We tested the hypothesis that perhexiline inhibits the enzyme carnitine palmitoyltransferase-1 (CPT-1), which controls access of long chain fatty acids to the mitochondrial site of beta-oxidation. Perhexiline produced a concentration-dependent inhibition of CPT-1 in rat cardiac and hepatic mitochondria in vitro, with half-maximal inhibition (IC50) at 77 and 148 mumol/L, respectively. Amiodarone, another drug with anti-anginal properties, also inhibited cardiac CPT-1 (IC50 = 228 mumol/L). The rank order of potency for inhibition was malonyl-CoA {\textgreater} 4-hydroxyphenylglyoxylate (HPG) = perhexiline {\textgreater} amiodarone = monohydroxy-perhexiline. Kinetic analysis revealed competitive inhibition of cardiac and hepatic CPT-1 by perhexiline with respect to palmitoyl-CoA but non-competitive inhibition with respect to carnitine. Curvilinear Dixon plots generated apparent inhibitory constant (Ki)" values for perhexiline which indicated a greater sensitivity of the cardiac than the hepatic enzyme to inhibition by perhexiline. Perhexiline inhibition of CPT-1 unlike that of malonyl-CoA and HPG was unaffected by pretreatment with the protease nagarse. These data establish for the first time that two agents with proven anti-anginal effects inhibit cardiac CPT-1. This action is likely to contribute to the anti-ischaemic effects of both perhexiline and amiodarone."
Journal of cardiovascular pharmacology 1995 mar
Direct evidence that perhexiline modifies myocardial substrate utilization from fatty acids to lactate.
Abstract
Abstract
Perhexiline maleate, originally classified as a calcium antagonist, is in use as an antianginal agent. The mechanism of its protective effect is unknown, but there is speculation that it involves a modification of myocardial substrate utilization, in which glycolytic sources are used rather than fatty acids. This hypothesis was tested by employing [13C]NMR isotopomer analysis to measure substrate selection in the working rat heart. Substrate utilization was measured from a mixture of substrates present at their physiological concentration, as follows: acetoacetate, glucose, lactate and long-chain fatty acids. Control perfusions were compared with those perfused with perhexiline. It was found that perhexiline increased lactate utilization, which reduced the extent of fatty acid and endogenous substrate oxidation. There was also a significant increase in cardiac output for a small and insignificant increase in oxygen consumption, which suggested an improvement in myocardial efficiency. Thus, it was confirmed by direct measurement that this drug does modify substrate oxidation, which suggests that further investigations of the role that this agent can play in the management of ischemic heart disease would be beneficial.
